O neurofibroma é a principal neoplasia benigna de bainha neural, podendo ser esporádico ou componente da síndrome da neurofibromatose do tipo 1. Essa síndrome é uma desordem autossômica dominante causada por um espectro de mutações envolvendo o gene da neurofibromatose 1 (NF1). Os principais achados da síndrome incluem: mancha “café com leite” na pele, neurofibromas na pele, neurofibroma plexiforme, defeitos ósseos, nódulos de Lish e tumores do sistema nervoso central. As lesões pigmentadas na neurofibromatose do tipo 1 apresentam bordas lisas, que lembram o estado da California; em contraste com as bordas irregulares, semelhantes a costa de Maine, da Síndrome de McCune-Albright. Lesão central de células gigantes dos ossos maxilares é também eventualmente encontrada.
A etiologia do neurofibroma está relacionada com a inativação bi-alélica do gene NF1, levando a perda completa de função do produto do gene, neurofibromina (NF1). Entretanto, estudos recentes mostram que o evento genético isoladamente não é suficiente para o surgimento da lesão. Sinais do microambiente inflamatório, envolvendo mastócitos, linfócitos, macrófagos, células dendríticas, bem como a interação das células de Schwann neoplásicas com axônios são necessários para o desenvolvimento do tumor. Estudos mais recentes sugerem que a perda de heterozigosidade no gene NF-1 em células tronco precursoras pode estar relacionada à patogênese da doença. De toda maneira, células com diferenciação para células Schwann representam o componente neoplásico do tumor.
Características clínicas
Embora o neurofibroma ocorra mais frequentemente na pele, não é incomum ser encontrado na boca. Ele pode ser localizado (cutâneo ou subcutâneo) ou difuso. A forma difusa é mais observada na região de cabeça e pescoço. O neurofibroma pode afetar jovens e adultos e ocorre principalmente no palato, língua e gengiva (Figura 1). Os pacientes com tumor, que apresentam a síndrome, geralmente, são mais jovens. A presença de lesões na língua, múltiplas ou do subtipo plexiforme deve levantar a suspeita de neurofibromatose do tipo 1.
A apresentação clínica do neurofibroma é de um nódulo ou tumor indolor, recoberto por uma mucosa de aparência normal e de crescimento lento. Embora raro, o tumor pode ocorrer dentro do osso, especialmente mandíbula, e leva a formação de uma imagem exibindo a dilatação fusiforme do canal mandibular (Figura 2).
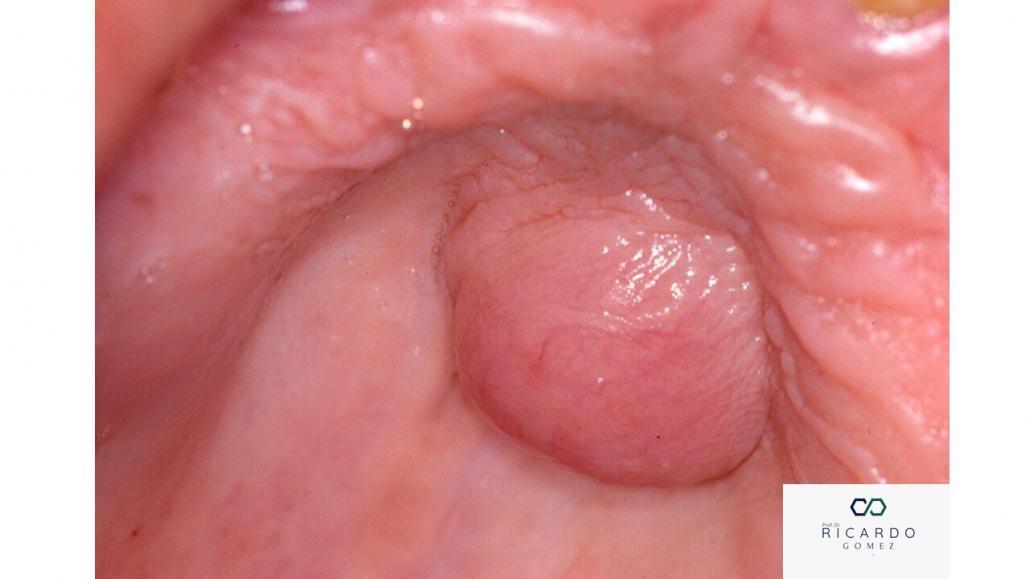
Figura 1- Imagem clínica do neurofibroma na porção anterior do palato duro.

Figura 2- Neurofibroma provocando dilatação fusiforme do canal mandibular.
Características histopatológicas
O neurofibroma é constituído por células de Schwann, células perineuriais, fibroblastos, mastócitos e por axônios mielinizados e não mielizados remanescentes, incluídos em uma matriz mixoide ou colagenosa. Apesar dos vários constituintes celulares, somente a subpopulação das células de Schwnn são consideradas neoplásicas. A lesão se apresenta bem delimitada, mas não encapsulada, composta por células fusiformes, contendo núcleos ondulados e com citoplasma escasso (Figura 3). Apesar dessas células serem de Schwann, elas são menores do que aquelas encontradas no Schwannoma. Essas células são separadas por feixes de fibras colágenas, podendo haver áreas mixoides. Diferente do padrão mais circunscrito encontrado no Schwannoma, o neurofibroma tende a infiltrar os tecidos adjacentes. Corpos de Pseudomeissnerian são encontrados principalmente nos casos difusos ou plexiformes.
Os principais subtipos histológicos são o neurofibroma antigo, celular, plexiforme e o neurofibroma atípico. No subtipo antigo poderemos encontrar alguns núcleos bizarros ou atípicos, sem a presença de outras alterações de malignidade. Ele não deve ser confundido com o neurofibroma atípico por não ser pré-maligno. Aumento da celularidade, na ausência de atividade mitótica e atipia, caracterizam a variante celular de neurofibroma. A variante histológica plexiforme apresenta aparência multinodular e frequentemente está associada com a neurofibromatose do tipo I. O neurofibroma atípico, também conhecida como neoplasia neurofibromatosa atípica de potencial biológico incerto, é caracterizado pela presença de duas ou mais das seguintes alterações: atipia celular, aumento da celularidade e perda da arquitetura do neurofibroma. As figuras de mitose devem se limitar até 3 por dez campos, em maior aumento, ou 1,5 /mm2. Esse subtipo é considerado pré-maligno ou em estágio inicial de malignidade e é encontrado principalmente em indivíduos portadores da síndrome.
No exame de imunohistoquímico do neurofibroma encontraremos positividade para as proteínas S-100, SOX10 e colágeno IV, que são marcadores de células de Schwann. Em razão da presença variável das células de Schwann, essa positividade não chega a ser tão extensa como no Schwannoma. Ocasionais células não neoplásicas positivas para EMA e CD34 são também observadas.
Figura 3: Imagem microscópica do neurofibroma mostrando células com núcleos ondulados.
Tratamento
O tratamento do neurofibroma é excisão cirúrgica. As recidivas são raras, bem como a transformação maligna. Com o diagnóstico da doença, a possibilidade da síndrome da neurofibromatose do tipo 1 deve ser investigada.
Leitura complementar:
1- Alotaiby FM, Fitzpatrick S, Upadhyaya J, Islam MN, Cohen D, Bhattacharyya I. Demographic, clinical and histopathological features of oral neural neoplasms: a retrospective study. Head Neck Pathol 2019;13:208-214.
2- Li S, Chen Z, Le LQ. New insights into the neurofibroma tumor cells of origin. Neurooncol Adv 2019;2(Suppl 1):i13-i22.
3- Miettinen MM, Antonescu CR, Fletcher CDM, Kim A, Lazar AJ, Quezado MM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10.
4- Thompson LDR, Koh SS, Lau SK. Sporadic neurofibroma of the tongue unassociated with neurofibromatosis type I: A clinicopathologic study of ten cases. Hed Neck Pathol 2019;14:374-380.
5- WHO Classification of Tumors Editorial Board. Soft Tissue and Bone Tumours. World Health Organization, 5 ed., 2020.
Consultório
Conheça nossos cursos